Changelog
Explanations of major changes to pixy are listed below. For up-to-date
info on minor versions and bugfixes, see the release notes on GitHub:
https://github.com/ksamuk/pixy/releases
pixy 2.2.3
This release contains the same analytical changes as the 2.2.2 tag, which
was released with its internal version strings left at 2.2.1: builds
from that tag report pixy 2.2.1 from pixy --version and in output
headers. 2.2.3 corrects the version strings only; if you are on 2.2.2,
there is no change in results, but --version will now agree with the
release you installed.
New features
wisp companion-tool input (``--wisp_bed``). Pass a wisp-format quantized callable-sites BED alongside a variants-only VCF; the per-window callable-site denominator for π, dxy, Watterson's θ, and Tajima's D is then sourced from the wisp mask rather than from invariant sites in the VCF, while FST continues to use only variant sites. This avoids the disk cost of an all-sites VCF for datasets where the callable-site pattern is already compact (see the companion tool at https://github.com/samuk-lab/wisp). Incompatible with
--gvcf.
Bug fixes
Tajima's D no longer collapses toward 0 with missing data (reinstates the fix for issue #160). The denominator of D is again evaluated once, at the mean number of observed alleles per site, as contributed by akihirao's
ThetaRecovand originally released in 2.0.0.beta10. That fix was inadvertently reverted in 2.1.0 while adding windowed Tajima's D aggregation, which needed a denominator that could be summed across window pieces and so rebuilt it as a per-observed-allele-count -class sum. Summing the square root of each class computessum(sqrt(v))where the correct form issqrt(sum(v)); sincesum(sqrt(v)) >= sqrt(sum(v)), the denominator was inflated as soon as missing data made the per-site observed-allele count ragged, shrinking |*D*| toward 0 and narrowing its distribution. Measured on a simulated growth model (true D ≈ −1.84): 2.2.1 reported −1.38 at 20% missing genotypes and −1.18 at 60%, versus −1.82 and −1.65 now. Data with no missing genotypes is unaffected — every site then has the same observed allele count, and the two forms coincide exactly. Aggregation is preserved and is now exact rather than approximate: the two denominator components are additive across window pieces, and the rounding to an integer mean happens once, on the pooled totals.The
--tajima_componentsoutput column has changed as a consequence. It is renamedtajima_d_s_counts→tajima_d_componentsand now holdsnsum=<int>,mut=<int>(the summed observed allele count and the total mutation count) rather thann:sobserved-allele-count classes, which are not sufficient to reconstruct the corrected denominator. Sum both fields, andno_sites, across the rows of a window to recomputetajima_d_stdevexactly.Sites fixed for the alternate allele are no longer counted as segregating. A site at which every sample is homozygous for the alternate allele has a single observed allele and so is not polymorphic, but
pixyup to 2.2.1 tallied one segregating site for every site with a non-zero alternate count, including these. This inflated Watterson's θ and depressed Tajima's D. This affects the default biallelic code path, not only--include_multiallelic_snps: θW and Tajima's D will change for any dataset containing alternate-fixed sites, which are common whenever the reference is diverged from the sampled population, or a population is fixed for a derived allele. π, dxy, and FST are unaffected. On biallelic inputpixynow agrees withscikit-allel'swatterson_thetaandtajima_d, which it did not before. Relatedly, a site with a single observed haploid genotype now contributes 0 rather than producing an infinite θW.Watterson's θ and Tajima's D now count mutations rather than segregating sites at multiallelic sites. Applies to
--include_multiallelic_snpsonly. A site with k observed alleles now contributes k − 1 mutations — Tajima'ss*, the minimum (parsimony) mutation count per site (Tajima 1996) — where it previously contributed 1 regardless of k. Watterson's estimator targets the number of mutations on the genealogy; counting sites is a shorthand that is exact only under strict infinite sites, and a multiallelic site is by definition where that shorthand breaks. The previous mismatch — multiallelic-aware π counting allelic differences while θW counted sites — biased Tajima's D upward, and the bias grew with the number of sampled lineages (ploidy × sample size). On biallelic data every k − 1 is 1, so the estimator is unchanged there. Multiallelic θW and Tajima's D now depart by design from implementations that count sites (scikit-allel,vcftools); this is worth stating in a methods section. Note that under recurrent mutations*is a parsimony minimum, so θW retains a small (~4%) downward bias that no model-free estimator can remove.
pixy 2.2.0
New features
Direct GVCF input. Pass
--gvcfto feed a joint-called GVCF (one in which runs of consecutive invariant positions are stored as block records withALT=<NON_REF>andINFO/END) directly topixy. Invariant blocks are expanded into per-site rows at read time, producing identical results to a fully-decompressed all-sites VCF without the intermediate file. See Arguments for the new--gvcfand--gvcf_max_block_sizeflags, and the GATK section of Generating invariant sites VCFs for how this lets you skip theGenotypeGVCFs --all-sitesstep. Incompatible with--wisp_bed; a clear error is raised if both are supplied or if--gvcfis set against a non-GVCF input.
pixy 2.0.0
pixy 2.0 is a major release that adds two new estimators, broadens
the range of organisms pixy can be used on, and introduces support
for multiallelic sites. The core π / dxy / FST
behaviour is preserved and remains backward-compatible with 1.x for
biallelic, diploid input.
If you use the new Watterson's θ or Tajima's D estimators, please cite the companion paper:
Bailey, N., Stevison, L., & Samuk, K. (2025). Correcting for bias in estimates of θW and Tajima's D from missing data in next-generation sequencing. Molecular Ecology Resources, e14104. https://doi.org/10.1111/1755-0998.14104
New features
Unbiased estimators of Watterson's θ and Tajima's *D*. Pass
--stats watterson_thetaand/or--stats tajima_dto compute them. Both correct for missing data the same waypixycorrects π and dxy. See Understanding pixy output for the output column reference.Multiallelic site support. Pass
--include_multiallelic_snpsto include sites with more than two alleles. Disabled by default (biallelic mode is slightly faster).Arbitrary and variable ploidy.
pixynow handles organisms of any ploidy, and ploidy may vary across chromosomes/contigs (so diploid autosomes alongside haploid sex chromosomes or organellar contigs work without splitting the VCF). Per-contig ploidy is inferred from the first record of each contig at startup. Note that the Weir & Cockerham (1984) FST estimator is only defined for diploid data; on non-diploid contigspixywill skip WC FST and emit a warning. Use--fst_type hudsonto compute FST on non-diploid contigs.CSI index support. Both
.tbi(tabix) and.csi(bcftools index) VCF indexes are accepted.Hudson's FST. Pass
--fst_type hudsonto use the Hudson (1992) / Bhatia et al. (2013) estimator instead of the default Weir & Cockerham (1984) estimator.
Bug fixes
--bed_filecoordinates are now interpreted as standard BED (0-based, half-open) rather than 1-based inclusive. Previously the rawchromStartwas treated as a 1-based inclusive start, which shifted the left edge of every window one base earlier than the user intended and inflated each window's length by one site. Users reusing existing BED files that were authored against the old behaviour should subtract 1 from eachchromStart.Multiallelic-site handling has been corrected. Previously, sites with more than two alleles could be counted incorrectly in FST computations.
Fixed the standard-deviation calculation used for Tajima's D.
Fixed dxy and FST for haploid input.
Fixed Watterson's θ for haploid input.
The check for invariant sites is now more permissive about VCF formatting and no longer false-positives on valid all-sites VCFs (#185).
Suppress noisy warnings from
scikit-allel(#183).Tabix index
mtimeis refreshed when needed to avoidindex older than datawarnings on some filesystems.
Project / packaging
Build system migrated to Poetry.
Strict static type-checking with mypy.
rufffor linting and formatting.Continuous integration runs the full test suite on every PR.
Python 3.10 through 3.14 are supported.
pixy 1.0.0
To coincide with the publication of the pixy manuscript, we're
very happy to announce the release of pixy version 1.0.0.
This was a major update to pixy and included a number of major
performance increases, new features, simplifications, and many minor
fixes. Note that this version contains breaking changes, and old
pipelines will need to be updated. We have also validated that the
estimates of π, dxy and FST produced by 1.0.0 are
identical to those of 0.95.02 (the version used in the manuscript).
Summary of major changes
All calculations are now much faster and natively parallelizable.
Memory usage vastly reduced.
BED and sites file support allows huge flexibility in windows / targeting sites of different classes of genomic elements.
Genotype filtration has been removed.
No change in the core summary statistics (π, dxy, FST) produced by
pixy.htslibis now a hard dependency, and must be installed separately.VCFs must be compressed with
bgzipand indexed withtabix(from htslib) before being used withpixy.
The performance increase and stability of numerical results are shown in the following plots:
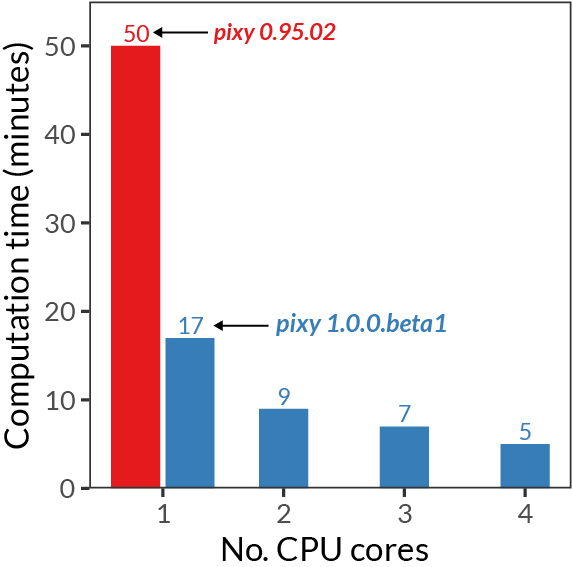
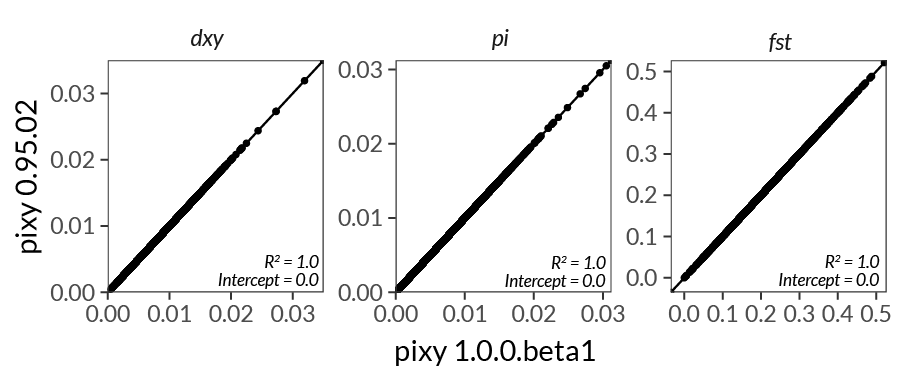
Detailed changelog
Major changes
pixycalculations can now be fully parallelized by specifying--n_cores [number of cores]at the command line.Implemented using the
multiprocessingmodule, which is now a hard dependency.Supported under both Linux and macOS (using fork and spawn modes respectively).
Many of the core computations have been vectorized with NumPy, resulting in significant performance gains.
Memory usage is now much lower, more intelligently handled, and configurable by the user via the
--chunk_sizeargument.Large windows (e.g. whole chromosomes) are dynamically split into chunks and reassembled after summarization.
Small windows are grouped into larger chunks to prevent I/O bottlenecks associated with frequently re-reading the source VCF.
New features
Support for BED files specifying windows over which to calculate π / dxy / FST. These windows can be heterogeneous in size, enabling precise matching of
pixyoutput with the output of other programs.Support for a tab-separated 'sites file' specifying sites (CHROM, POS) where summary statistics should be exclusively calculated. This also enables, for example, estimates of π using only 4-fold degenerate sites or for a particular class of genes.
Basic support for site-level statistics (1 bp scale, though much slower than windowed statistics).
Removed features
pixyno longer makes use of a Zarr database for storing on-disk intermediate genotype information. We instead now perform random access of the VCF via tabix from htslib as implemented inscikit-allel. As such, htslib is now a hard dependency. We think tabix is a much more flexible system for many datasets, and the performance differences are negligible (and offset by the new performance features in v1.0). VCFs will need to be compressed withbgzipand indexed withtabixbefore usingpixy.Other than requiring all variants to be biallelic SNPs,
pixyno longer performs filtration of any kind. We decided that filtration was outside the scope of the functionality we wantedpixyto have. There are already many excellent tools that perform filtration, and pre-filtering creates a filtered VCF that can be used for other analyses. We now strongly recommend that users pre-filter their invariant sites VCFs using VCFtools and/or BCFtools. We provide an example shell script with this functionality (retaining invariant sites as required) as a template for users to edit for their needs.
Minor updates
The pre-calculation checks performed by
pixyare now more extensive and systematic.The method for calculating the number of valid sites has been slightly adjusted to be more accurate (this was calculated independently of the π / dxy / FST statistics).
We've refactored and restructured much of the code, with a focus on increased functionalization. This should make community contributions and future updates much easier.
To reduce confusion, output prefix and output folder are now separate arguments.
The documentation for
pixyhas been extensively updated to reflect the new changes in version 1.0.0.
Other bugfixes
Total computation time is now properly displayed.
For FST: regions with no variant sites will now have
NAin the output file, instead of not being represented.